La contraction musculaire
Vidéo en bas de page ^^
I L’ATP, source d’énergie de la cellule musculaire
La contraction d’un muscle squelettique, à l’origine d’un mouvement volontaire, nécessite un travail cellulaire. Les cellules musculaires effectuent ce travail en transformant l’énergie chimique des molécules organiques en énergie mécanique. L’ATP est un intermédiaire dans cette conversion.
Document 1 : Molécule d’ATP
v
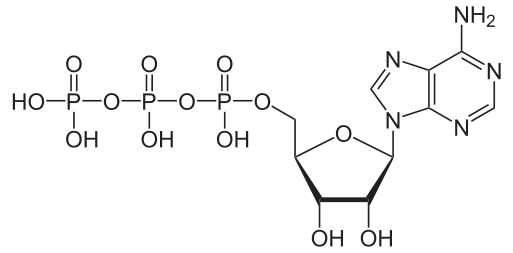
Adenosintriphosphat_protoniert.svg par NEUROtiker propre travail, via Wikimédia Commons, domaine publique, https://commons.wikimedia.org/wiki/File:Adenosintriphosphat_protoniert.svg
L’ATP ou adénosine triphosphate est composée d’une base azotée (l’adénine), d’un sucre à 5 carbones (ribose) et de trois groupements phosphate (H3PO4). L’hydrolyse de l’ATP (de la dernière liaison phosphate) en un ADP (adénosine diphosphate) et un phosphate inorganique Pi, libère de l’énergie à raison de 30.5 kJ.mol-1 : on parle de réaction exergonique. L’ATP stocke l’énergie en créant une liaison phosphate lors de sa formation par phosphorylation : c’est une réaction endergonique.
Document 2 : Hydrolyse de l’ATP et Phosphorylation de l’ADP.
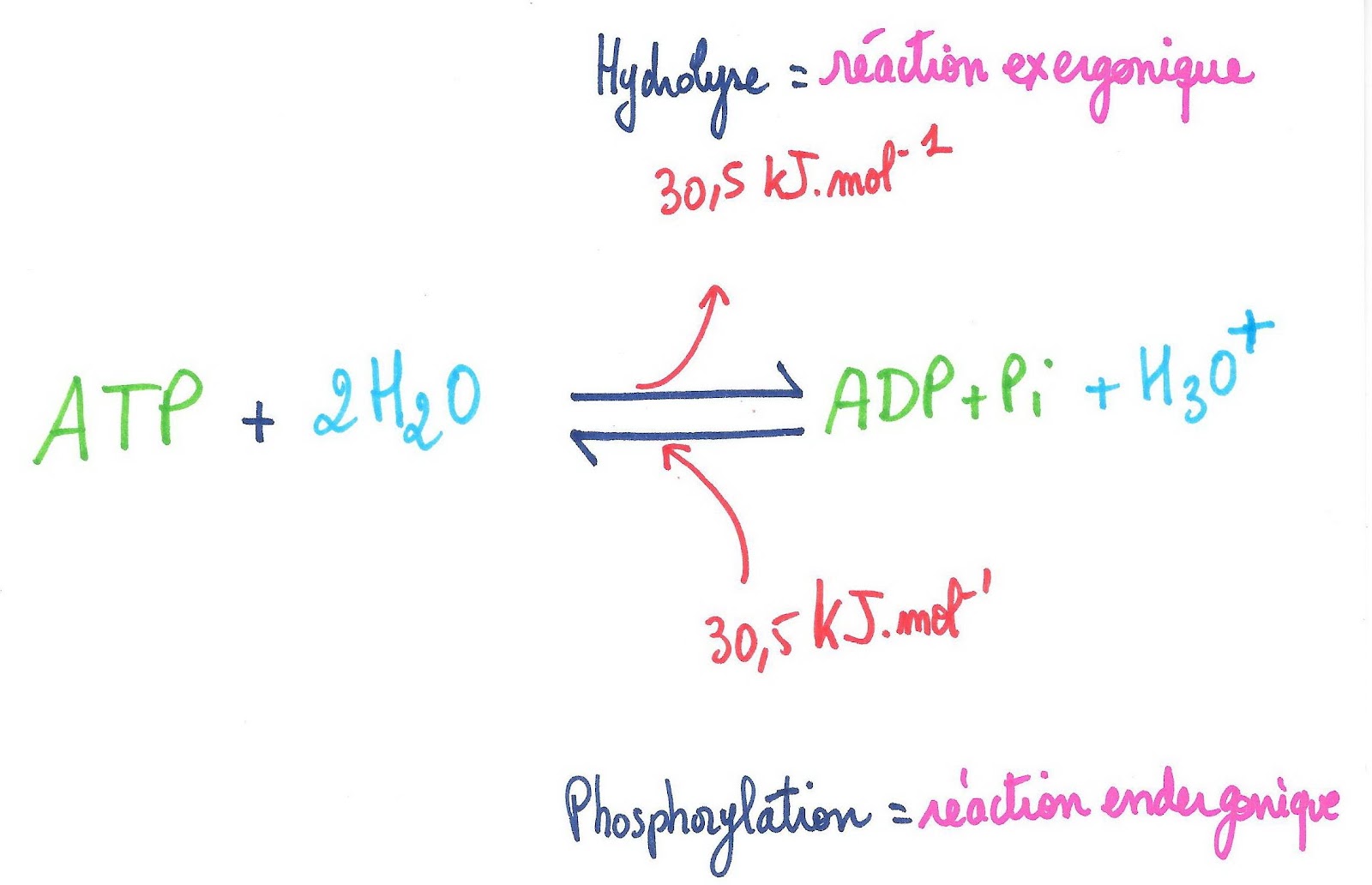
©RS.2021
Ainsi, le corps humain ne contient à chaque instant qu'environ 250 g d'ATP mais consomme et régénère chaque jour l’équivalent de son propre poids en cette molécule énergétique. L’énergie chimique libérée par l’hydrolyse de l’ATP permet en partie la production d’une énergie mécanique et le reste est dissipé sous forme de chaleur.
II Mécanismes moléculaires de la contraction
Document 3 : Étapes moléculaires de la contraction musculaire

Querbrückenzyklus 1.png, Querbrückenzyklus_2, Querbrückenzyklus_3, Querbrückenzyklus_4, Originaux téléversés par Moralapostel sur Wikipédia allemand, CC-BY-SA-3.0-migrated, modifiés par Sandra Rivière
https://commons.wikimedia.org/wiki/File:Querbr%C3%BCckenzyklus_1.png?uselang=fr, https://fr.wikipedia.org/wiki/Fichier:Querbr%C3%BCckenzyklus_2.png
https://commons.wikimedia.org/wiki/File:Querbr%C3%BCckenzyklus_3.png
https://commons.wikimedia.org/wiki/File:Querbr%C3%BCckenzyklus_4.png
La tête de myosine passe son temps à se décrocher et à se raccrocher au filament d’actine :
- Les têtes de myosine viennent de fixer chacune une molécule d’ATP.
- Cette fixation provoque la séparation des têtes de myosine du myofilament d’actine avec lequel elles formaient un angle de 45° environ.
- Les têtes de myosine hydrolysent l’ATP en ADP et Pi.
- Cette hydrolyse leur permet de changer de conformation et de se positionner perpendiculairement à l’axe du myofilament d’actine.
- Les têtes de myosine porteuses d’ADP et de Pi se lient aux molécules d’actine (formation des complexes acto-myosine).
- Les têtes de myosine libèrent l’ADP et le Pi.
- Cela provoque un changement de conformation spatiale de celles-ci : elles pivotent de 45° faisant glisser les myofilament d’actine vers le centre du sarcomère.
Le raccourcissement des sarcomères est donc dû à un cycle de liaison-dissociation entre actine et myosine associé à des changements de conformation des têtes de myosine. Ce cycle se produit tant que de l’ATP est disponible et que la concentration en Ca2+ est élevée (au moins 1 μmol.L-1), celui-ci étant nécessaire pour démasquer les sites de liaison situés dans les molécules d'actine. En effet, le filament d’actine est maintenu stable par 2 protéines : la tropomyosine très longue qui enserre les filaments d’actine et des complexes de troponine. Ces derniers masquent les sites de liaison de la myosine avec l’actine. Le potentiel d’action musculaire initié au niveau de la plaque motrice se déplace de proche en proche dans la membrane plasmique par l’ouverture de canaux sodiques voltage-dépendants distribuant la dépolarisation aux tubules T qui se dépolarisent et induisent une libération d’ions calcium du réticulum sarcoplasmique vers le sarcoplasme de la fibre musculaire. Le calcium libéré va se fixer sur la troponine induisant un changement de position de l’ensemble troponine-tropomyosine et donc la libération des sites de fixations.
Document 4 : Troponine et site de fixation de la myosine
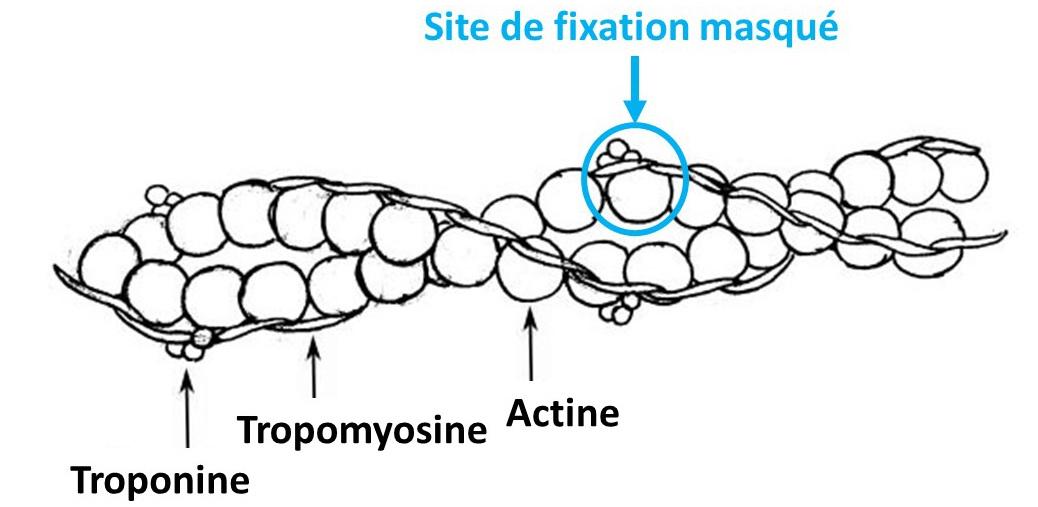
Thin_filament, Rama . Modifié par l' utilisateur: Raul654, via Wikimédia Commons, CC-BY-SA-3.0-migré, modifié par Sandra Rivière, https://commons.wikimedia.org/wiki/File:Thin_filament.jpg
III La myopathie de Duchenne, une anomalie touchant les muscles
A/ Symptômes
La myopathie de Duchenne est une maladie qui touche essentiellement les garçons. C’est une maladie génétique provoquant une dégénérescence musculaire. L’espérance de vie des personnes atteintes est de 30 ans.
La myopathie de Duchenne est caractérisée par un dysfonctionnement de tous les muscles (striés squelettiques, strié cardiaque et lisses) conduisant à une hypotrophie musculaire, c'est-à-dire à un retard de croissance des muscles.
Cela conduit à un apprentissage retardé de la marche, des chutes nombreuses et des difficultés pour se relever. Il y a une perte de motricité vers l’âge de 10 ans. Le myocarde est aussi touché (atrophie après 3 ans), comme les muscles lisses impactant le fonctionnement digestif. L’affaiblissement des capacités ventilatoires (diminution du volume inspiratoire) et cardiaque est à l’origine d’un diagnostic peu favorable.
Document 5 : Mesure de la force musculaire au fur et à mesure des cycles de contraction chez 2 lots de souris

©RS.2021
Des études chez des souris atteintes montrent une fatigabilité musculaire : au bout de six cycles de contraction, le muscle ne se contracte plus chez les souris mutées, alors que chez les souris sauvages témoins, le muscle se contracte encore à plus de 60% de sa force initiale. Les symptômes de la myopathie de Duchenne sont dus à une fragilité des muscles du corps.
Une étude histologique montre que les cellules musculaires sont remplacées par des cellules graisseuses.
Document 6 : Coupe histologique de mollet montrant un remplacement des fibres musculaires par des cellules adipeuses.
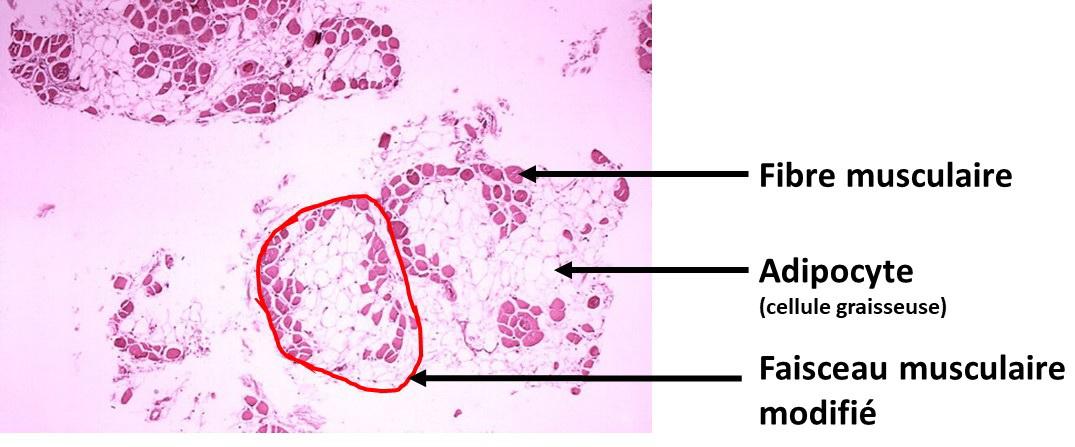
Duchenne-muscular-dystrophy par Dr Edwin P. Ewing, Jr. Via wikimedia commons, autorisation PD-USGov-HHS-CDC, domaine publique, https://commons.wikimedia.org/wiki/File:Duchenne-muscular-dystrophy.jpg
Il faut donc comprendre le mécanisme à l’origine de la disparition des fibres musculaires.
B/ Origine de l’insuffisance musculaire
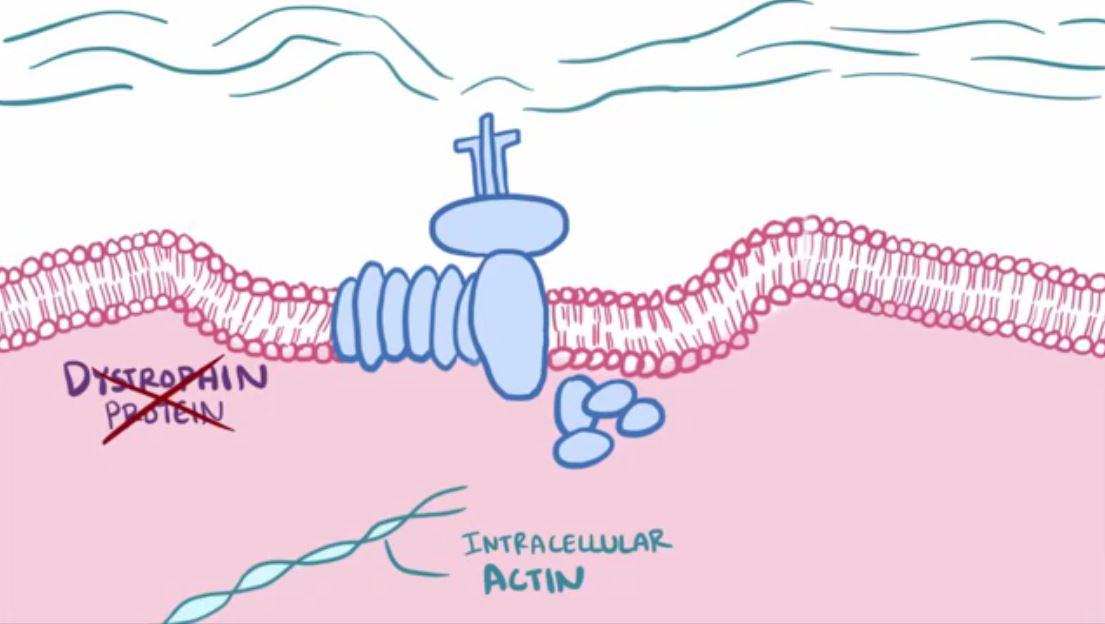
Des études histologiques et moléculaires poussées montrent que la myopathie de Duchenne est due à un déficit en une protéine du cytosquelette : la dystrophine. Cette protéine fait le lien entre la matrice extracellulaire et les myofibrilles des cellules musculaires.
Document 7 : organisation du cytosquelette et matrice extracellulaire

Dystrophie musculaire de Duchenne et Becker.webm, par Osmose via Wikimédia Commons, CC-BY-SA-4.0, https://commons.wikimedia.org/wiki/File:Duchenne_and_Becker_muscular_dystrophy.webm
En effet, les fibres de collagène de la matrice extracellulaire sont reliées à un complexe protéique membranaire lui-même relié au cytosquelette situé sous la membrane plasmique. Ces protéines de cytosquelette sont des filaments de dystrophine eux-mêmes associés à des filaments d’actines reliés aux myofibrilles. Dystrophine et actine servent donc de cytosquelette interne permettant le soutien des cellules musculaires. Cela permet de maintenir une cohésion entre le cytosquelette et la matrice extracellulaire, assurant un mouvement coordonné de l’ensemble des cellules du muscle à chaque cycle de contraction.
Document 8 : Faisceau de cellules musculaires observées au microscope à fluorescence chez un individu atteint ou non de DMD

MuscularDystrophy, par Cbenner12 via wikimedia commons, CC-BY-SA-3.0
https://commons.wikimedia.org/wiki/File:MuscularDystrophy.png
Une étude histologique par fluorescence montre que la dystrophine (en vert) est présente au niveau de toutes les membranes plasmiques des cellules musculaires (myocytes, en noir) d’individus sains témoins alors qu’elle a disparu sur les membranes des myocytes des myopathes.
Document 9 : Organisation du cytosquelette et matrice extracellulaire dans le cas de la DMD
Dystrophie musculaire de Duchenne et Becker.webm, par Osmose via Wikimédia Commons, CC-BY-SA-4.0, https://commons.wikimedia.org/wiki/File:Duchenne_and_Becker_muscular_dystrophy.webm
Ainsi chez les individus myopathes, l’absence de dystrophine entraîne une perturbation du lien entre le cytosquelette, la membrane et la matrice extracellulaire, ce qui abîme les cellules musculaires à chaque contraction. Leur membrane se déchire laissant s’échapper des enzymes de type créatine kinase et entrer du Ca2+. Ces échanges finissent par tuer la cellule.
Document 10 : Conséquences de la déchirure de la membrane plasmique dans le cas d’une DMD.

Dystrophie musculaire de Duchenne et Becker.webm, par Osmose via Wikimédia Commons, CC-BY-SA-4.0, https://commons.wikimedia.org/wiki/File:Duchenne_and_Becker_muscular_dystrophy.webm
La dystrophine étant une protéine, cette maladie est donc une maladie génétique liée au dysfonctionnement de son gène.
C/ Une maladie héréditaire
Le mode de transmission de cette maladie génétique est dit récessif et lié au gène DMD du chromosome X. Cela signifie que la maladie se déclare chez les individus n'ayant pas d'allèle fonctionnel pour ce gène. Les hommes n'ayant qu'un seul chromosome X, ils ne possèdent qu'un seul allèle : tous les hommes porteurs d'une mutation délétère de ce gène sont donc atteints. Les femmes ont deux chromosomes X . Les femmes portant une mutation ne sont pas atteintes car elles possèdent un deuxième allèle non muté : elles sont dites « porteuses-saines ». Pour qu’une femme soit atteinte, elle doit donc posséder deux exemplaires de l’allèle muté.
L’espérance de vie étant faible, peu d’individus atteints transmettent l’allèle muté. La transmission se fait donc principalement par les individus hétérozygotes.
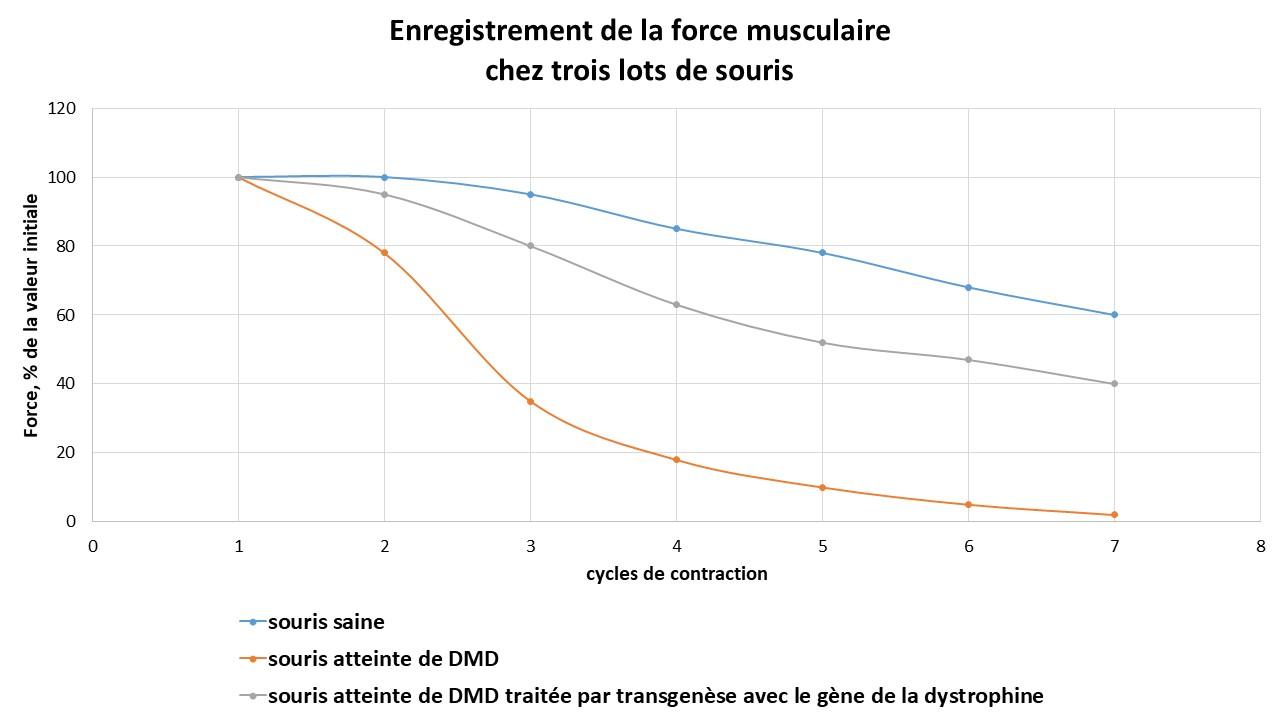
La transgénèse serait un espoir thérapeutique. Des expériences sur des souris montrent que le remplacement du gène non fonctionnel améliore la réponse musculaire.
Document 11 : Mesure de la force musculaire au fur et à mesure des cycles de contraction chez 3 lots de souris
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
©RS.2021
Contraction musculaire - SVT - SANTÉ Term spé #8 - Mathrix
Date de dernière mise à jour : 25/06/2021