Transmission de maladies génétiques
I Étude génétique d’une famille
A/ Étude d’un arbre généalogique d’une famille touchée par l’hémophilie
En génétique, l’arbre généalogique est une figure représentant un arbre dont les ramifications montrent la filiation des diverses branches d’une même famille. Les générations sont identifiées par des numéros exprimés en chiffres romains et les individus d’une même génération sont répertoriés par des numéros en chiffres arabes. Ainsi l’identification d’un individu se fait par une combinaison d’un chiffre romain et d’un chiffre arabe permettant de le localiser dans l’arbre. Les individus de sexe féminin sont représentés par des cercles et les individus de sexe masculin sont représentés par des carrés. Les individus malades ont leur symbole noirci. Un individu décédé est barré.
Document 1 : Exemple d’arbre généalogique de génétique montrant la transmission d’un trouble du langage.

Source : Pedigree KE-Family 01-fr.png par Kuebi = Armin Kübelbeck via Wikimédia Commons, CC-BY-SA-3.0, https://commons.wikimedia.org/wiki/File:KE-Family_pedigree_01-fr.png
Dans le cas de l’étude d’une maladie génétique, l’étude de l’arbre généalogique permettra dans un premier temps de démontrer si l’allèle responsable de la maladie est récessif ou dominant puis de définir le mode de transmission des gènes, à savoir si l’allèle est porté par un chromosome sexuel ou pas.
Afin de prouver la dominance ou la récessivité de l’allèle étudié, il faut sélectionner un individu malade et analyser le phénotype de ses parents. Si les deux parents sont sains et que cette situation se reproduit plusieurs fois dans la famille, alors l’allèle responsable de la maladie est récessif.
Il faut ensuite déterminer quelle catégorie de chromosome porte le gène responsable de la maladie. Il faut dans un premier temps traiter l’éventualité qui soit porté par un chromosome sexuel. Le chromosome Y n’étant porté que par les hommes, il est assez facile de déterminer son implication dans le mode de transmission. En effet, si le chromosome Y porte bien l’allèle responsable de la maladie alors un père malade ne peut avoir que des fils malades. De plus à partir du moment où il y a une femme malade dans la famille, la femme n’ayant pas de chromosome Y, l’allèle malade ne peut pas être sur le chromosome Y.
Dans le cas où le rôle du chromosome Y aurait été écarté, il convient de vérifier l’implication du chromosome X. Dans le cas où une majorité des malades sont des hommes et que l’allèle n’est pas porté par Y, on soupçonne que celui-ci soit porté par X. On va alors vérifier que toute fille malade a un père malade (elle n’a pu hériter de lui que de son X porteur de l’allèle malade) et que toute mère malade a un fils malade (comme il est XY il a reçu forcément l’allèle malade par le X de sa mère). Ce n’est pas une preuve absolue, mais il y a de très fortes chances que dans ce cas l’allèle responsable de la maladie soit sur le chromosome X. Il convient alors de vérifier tous les croisements de l’arbre.
On peut citer l’exemple l’hémophilie, maladie génétique dont la transmission est dite « récessive liée à X ».
Document 2 : Arbre généalogique d’une famille atteinte d’hémophilie

Les individus II1, II4, et III4 sont malades mais avec des parents sains. Cela signifie que les parents sont porteurs sains puisqu’ils transmettent la maladie sans être eux-mêmes malades. Ils ont donc un allèle sain et un allèle malade : ils sont hétérozygotes pour ce gène. L’allèle malade est donc récessif.
Il faut ensuite déterminer si cette maladie est autosomale ou liée à Y ou X, c'est-à-dire portée par un autosome et non par un gonosome (chromosome sexuel) ou au contraire porté par l’un des deux (Y ou X).
On remarque que l’individu III2 est malade or c’est une femme. On peut donc affirmer que l’allèle muté n’est pas porté par Y. Il est alors porté soit par un autosome soit par X.
Cette femme III2 a un père malade et elle engendre deux garçons malades. Ces derniers ayant reçu Y de leur père, ils ont forcément reçu X portant l’allèle muté de leur mère. Si la maladie avait été autosomale récessive en raison du brassage inter-chromosomique, il y aurait autant de chances d’avoir un garçon qu'une fille atteinte et le nombre de filles et de garçons atteints dans cette famille serait presque identique. Ce n’est pas le cas ici, on peut donc en conclure que l’allèle malade est bien porté par X.
Une fois le mode de transmission trouvé, il convient de le vérifier dans tout l’arbre.
Tous les hommes sains ont un X normal (et un Y) : I1, I4, I6, II2, II5, III1, IV4.
Les filles saines issues de père ou de mère malades sont porteuses (donc avec un X normal et un X muté) : III3 et IV2
Les mères saines ayant un fils ou une fille malade sont porteuses (donc avec un X normal et un X muté) : I2, I3, II3, II6
Tous les hommes malades ont un X muté (et un Y) : II1, II4, III4, IV1 et IV3
Individu I5 : Sachant que sa fille II6 est porteuse et que le père I6 est sain, alors I5 doit nécessairement être porteuse.
II7 et III5 : Il est impossible de déterminer si elles sont porteuses ou saine. En effet, avec un père sain et une mère porteuse, il y a donc une chance sur deux qu’elles soient saines, et de même pour qu’elles soient porteuses.
B/ Étude d’un arbre généalogique d’une famille touchée par le syndrome de Li-Fraumeni
Le syndrome de Li-Fraumeni est un syndrome rare de prédisposition au cancer caractérisé par l’apparition précoce de plusieurs cancers primitifs tels que le cancer du sein, le sarcome des os et des tissus mous ou encore des tumeurs cérébrales.
Il se définit par l’observation d’un sarcome chez un sujet atteint de moins de 45 ans, apparenté au premier degré à une personne ayant eu un cancer de n’importe quel type avant 45 ans, ou au deuxième degré à une personne ayant eu un cancer ou un sarcome à moins de 45 ans. C’est un cas rare de cancer héréditaire.
Dans la famille suivante la mère II2 a eu un cancer du sein à 30 ans et son fils III2 a eu un sarcome à 8 ans.
Document 3 : Arbre généalogique d’une famille atteinte du syndrome de Li-Fraumeni

L’arbre suggère une transmission héréditaire car il montre que 2 individus étroitement apparentés sont atteints d’un cancer précoce. Or le syndrome de Li-Fraumeni se définit par un sarcome chez un sujet de moins de 45 ans (ce qui est le cas de l’individu III2 qui a un sarcome à 8 ans) et par un lien de parenté au premier degré à une personne ayant eu un cancer à moins de 45 ans, ce qui est le cas de l’individu II2 (sa mère qui a eu un cancer à 30 ans). On pourrait donc bien parler de maladie héréditaire et non de maladie provenant d’une mutation somatique apparue chez les individus comme dans la plupart des cancers.
Un cancer correspond à une multiplication anarchique de cellules. Il est donc logique de soupçonner, dans le caryotype des cellules cancéreuses, l’existence d’une mutation du gène codant pour la protéine p53 régulatrice du cycle cellulaire. Pour en être sûr, il suffit de comparer la séquence des allèles responsables de la production de la protéine p53 dans les cellules somatiques et dans les cellules cancéreuses d’un individu malade. Si l’allèle défectueux n’est observé que dans les cellules cancéreuses alors on pourra affirmer que le cancer est dû à une mutation d’une cellule somatique et à ce moment-là la forte proportion de cancer dans cette famille pouvant faire penser à une maladie héréditaire est un pur hasard. Si au contraire l’allèle défectueux est observé dans toutes les cellules de l’organisme alors cela signifie qu’il était présent dès la cellule-œuf et que l’individu en a donc hérité de ses parents.
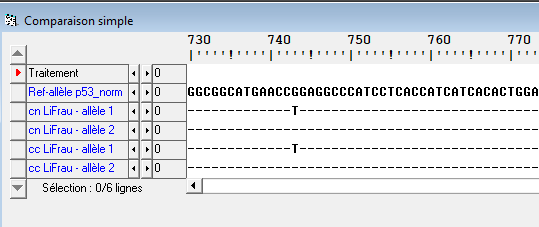
L’étude sous Anagène des allèles du gène p53 présents dans les cellules cancéreuses (cc) de l’individu II2 atteinte d’un cancer et les allèles du gène p53 présents dans ses cellules somatiques non cancéreuses (cn) révèlent que les deux catégories de cellules possèdent un allèle normal et un allèle muté en position 743. Comme la mutation est présente à la fois dans les cellules somatiques et dans les cellules cancéreuses, alors on peut affirmer que la mutation est héréditaire.
Document 4 : Comparaison des séquences des allèles du gène codant pour la protéine p53 sous anagène pour les cellules somatiques normales et pour les cellules cancéreuses de l’individu II2
Ainsi les parents I1 et I2 ont certainement transmis l’allèle muté à l’individu II2. Ils sont cependant sains. On peut donc supposer que l’allèle normal est dominant sur l’allèle malade et que ces 2 parents doivent porter cette mutation c’est-à-dire être hétérozygotes.
La comparaison sous Anagène des allèles portés par les parents I1 et I2 montre qu’ils ne possèdent pas d’allèle muté. Ils sont homozygotes pour l’allèle normal.
Document 5: Comparaison des séquences des allèles du gène codant pour la protéine p53 sous anagène pour les parents I1 et I2

On peut donc ainsi affirmer que l’allèle muté transmis par l’individu II2 à son fils résulte probablement d’une mutation ayant eu lieu dans une cellule germinale d’un de ses deux parents I1 ou I2. Il est également envisageable que la mutation ait eu lieu dans la cellule œuf à l’origine de l’individu II2 avant la première division de celle-ci.
On parle de mutation de novo, c'est-à-dire une mutation d’un gène apparaissant chez un individu alors qu’aucun de ses 2 parents ne la possède dans son patrimoine génétique.
II Maladie génétique et analyse prédictive, exemple de la mucoviscidose.
La mucoviscidose est une maladie génétique dite « autosomique récessive », transmise par 2 parents porteurs sains : chacun d’eux transmet un allèle muté du gène codant pour la protéine CFTR (Cystic fibrosis transmembrane conductance regulator), présent au niveau du chromosome 7. La protéine CFTR est une protéine présente dans la membrane des cellules de différentes muqueuses : respiratoire, digestive… Elle fonctionne comme un canal qui permet l’échange d’ions chlorure entre l’intérieur et l’extérieur de la cellule. Le fort taux d’ion chlorure crée un appel d’eau qui va fluidifier le mucus. Chez un individu atteint de mucoviscidose, ces protéines ne sont pas insérées dans la membrane plasmique apicale, elles sont stockées dans le cytoplasme pour être au final détruites. De ce fait, les cellules épithéliales ne peuvent exporter les ions chlorure dans la lumière des bronches : le mucus s’épaissit. Les cils de ces cellules épithéliales n’arrivent pas à évacuer ce mucus : il stagne, obstrue les bronches, empêchant le passage de l’air. Les apports en dioxygène sont faibles et l’individu se fatigue. De plus un mucus épais favorise la prolifération de micro-organismes. Un mécanisme similaire s’observe dans les canaux pancréatiques.
Document 6 : Phénotype de la mucoviscidose à différentes échelles
|
|
|
|
|
|
|
|
La protéine CFTR est formée d'une chaîne unique de 1480 acides aminés. La structure tridimensionnelle de la protéine montre une portion constituée de motifs en hélice et correspondant aux domaines transmembranaires (MSD1 et MSD2 - Membrane Spaning Domain). Ils sont surmontés par deux domaines intracellulaires capables de se lier à l'ATP (adénosine tri-phosphate riche en énergie), appelés NBD1 et NBD2 (Nucleotide Binding Domain). Enfin, une région régulatrice (R) leur est juxtaposée. L'hydrolyse de l'ATP au niveau des domaines NBD intracellulaires déclenche un changement de conformation des domaines transmembranaires, ce qui permet l'ouverture du canal et le passage des ions chlorure dans le milieu extérieur.
Document 7 : Structure simplifié du canal CFTR
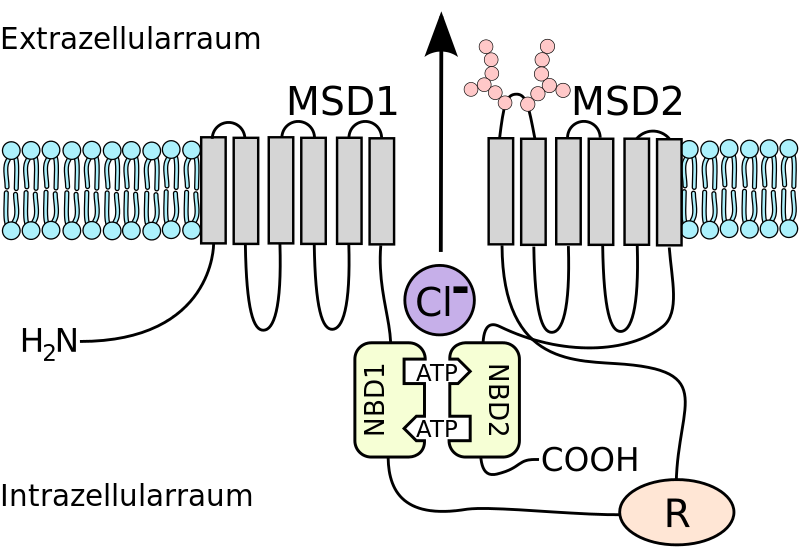
Source : schéma de structure de la protéine CFTR 02.svg, Par Kuebi = Armin Kübelbeck via Wikimédia Commons, CC-BY-SA-3.0 https://commons.wikimedia.org/wiki/File:CFTR_protein_structure_scheme_02.svg
Document 8 : Localisation du gène CFTR sur le chromosome 7
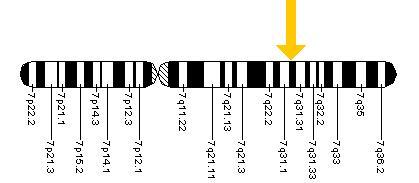
Source : Localisation du gène CFTR : Cftr.jpeg, domaine public via Wikimédia Commons, https://ar.m.wikipedia.org/wiki/%D9%85%D9%84%D9%81:Cftr.jpeg
Il existe plusieurs mutations de la partie codante du gène. La comparaison de l’allèle sain CFTR-CDS.Adn avec l’allèle muté CFTR-R553X.adn révèle une substitution d’une cytosine par une thymine en position 1657 provoquant l’apparition d’un codon stop. C’est une mutation non-sens. La protéine n’est donc pas synthétisée.
Document 9 : Comparaison sous Anagène de la séquence en nucléotides de l’allèle CFTR-CDS et de l’allèle CFTR-R553X

Document 10 : Comparaison sous Anagène de la séquence en acides aminés de la protéine CFTR-CDS et de la protéine CFTR-R553X

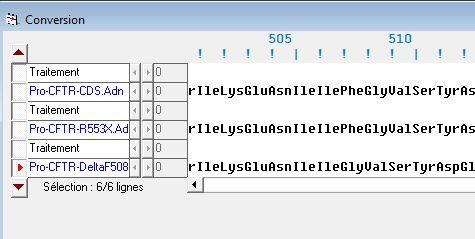
La comparaison de l’allèle sain CFTR-CDS.Adn avec l’allèle muté CFTR-DeltaF508.Adn révèle une délétion entre des positions 1522, 1523 et 1524 des nucléotides. Il y a alors disparition de 3 thymines provoquant la disparition d’une phénylalanine en position 508. La délétion se fait à cheval sur deux codons : perte d’une base en position trois du premier codon et perte des deux premières bases du codon suivant. L’association des bases restantes code pour le même acide aminé que le premier codon original. Il y a donc juste une perte de la phénylalanine et un décalage dans la séquence peptidique. C’est une mutation faux-sens. Cela provoque une mauvaise conformation spatiale de la protéine expliquant son absence de migration dans la membrane plasmique. Cette mutation est la plus fréquente dans la population : 1 individu sur 34 est porteur de l’allèle muté CFTR-deltaF508.
Document 11 : Comparaison sous Anagène de la séquence en nucléotides de l’allèle CFTR-CDS et de l’allèle CFTR-DeltaF508

Document 12 : Comparaison sous Anagène de la séquence en acides aminés de la protéine CFTR-CDS et de la protéine CFTR-DeltaF508: disparition de la phénylalanine en position 508 et décalage du cadre de lecture vers la gauche.
La mucoviscidose est systématiquement diagnostiquée dès la naissance depuis 2002. Il existe plusieurs diagnostics possibles. Le premier est réalisé au troisième jour après la naissance. Il consiste à prélever un peu de sang au talon du nouveau-né recueilli sur un morceau de papier buvard (test de Guthrie). Il suffit ensuite de doser la trypsine immunoréactive dans le sang du nouveau-né. Produite par le pancréas et intervenant dans les sucs digestifs, elle est normalement présente en faible quantité dans le sang mais en cas d’obstruction des voies pancréatiques sa concentration augmente fortement. Ce n’est pas forcément synonyme de mucoviscidose mais ce test oriente le diagnostic. Une recherche de mutation du gène CFTR est alors enclenchée à partir du même échantillon sanguin. Un test biologique est systématiquement réalisé. C’est le test dit « de la sueur » qui consiste à doser les ions chlorures après avoir provoqué la sudation du patient : leur taux sera anormalement élevé chez les patients atteints de mucoviscidose.
Enfin il est possible de réaliser l’analyse génétique de la famille quand dans une famille des cas sont recensés. Ceci permet une prédiction du risque pour les 2 personnes d’un couple d’être malade ou d’être porteur sain. Si les 2 parents d’un fœtus sont tous deux hétérozygotes, ils auront 1 risque sur 4 d’avoir un enfant malade et 2 risques sur 4 qu’il soit porteur sain. Le couple bénéficiera d’un diagnostic prénatal qui consiste en une analyse génétique des cellules fœtales récupérées par amniocentèse dans le liquide amniotique.
Document 12 : Transmission autosomique récessive et risque d’être malade
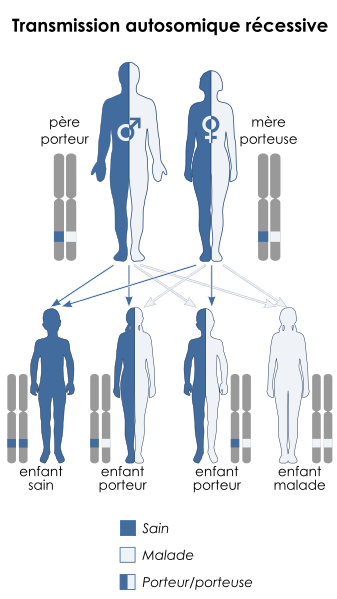
Source : Autosomique récessif - fr.svg , Kashmiri , basé sur des travaux antérieurs de Domaina
, via Wikimédia CommonsCC-BY-SA-3.0, https://commons.wikimedia.org/wiki/File:Autosomal_recessive_-_fr.svg
Document 13 : Principe de l’amniocentèse
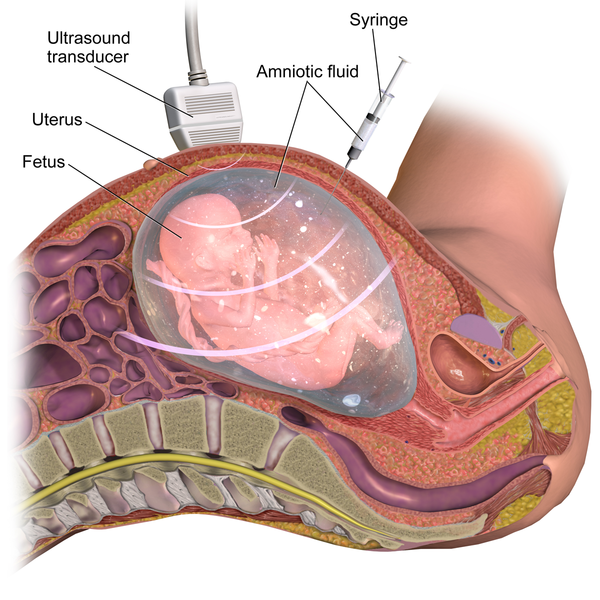
{kind=link}
Source : Amniocentesis.png par BruceBlaus, CC-BY-SA-4.0, via Wikimédia Commons, https://commons.wikimedia.org/wiki/File:Amniocentesis.png
Maladies génétiques- SVT - LA VIE Term spé #7 - Mathrix
Date de dernière mise à jour : 26/05/2021