Lignée clonale
vidéo en bas de page ^^
I La durée de vie des cellules est normalement définie
Un clone est donc une masse de cellules issues de divisions successives à partir d’une cellule.
Le nombre de divisions cellulaires est contrôlé et non indéfini. En effet plus une cellule est issue d’un grand nombre de divisions, plus elle est susceptible d’accumuler des mutations. Pour éviter l’apparition d’un clone muté, le nombre de divisions est contrôlé par des séquences d’ADN répétitives ne contenant pas de gènes et appelé « télomères ». Ce mot vient du grec ancien télos (« fin ») et du grec ancien méros (« partie »). C’est donc la partie située à la fin des chromosomes, par opposition au centromère qui se trouve au centre de ceux-ci.
À chaque fois que la cellule recopie son ADN, avant de se diviser, elle perd un petit bout de télomère (comme une photocopieuse qui rognerait les marges du document original). Ainsi au bout d’un certain nombre de divisions, les télomères n’existent plus ce qui stoppe la division cellulaire. La cellule devient sénescente (elle vieillit) et fonctionne moins bien. Elle finit par mourir. On parle d’apoptose. Ce processus participe au vieillissement de l’organisme.
Document 1 : Télomères et divisions
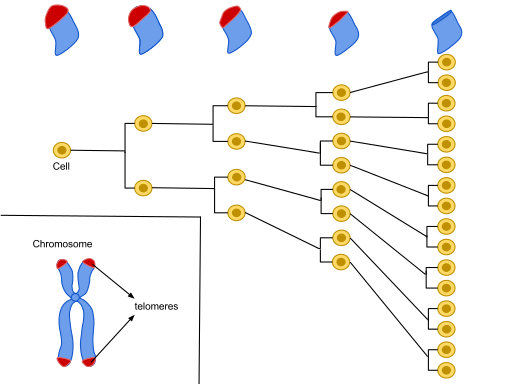
Sources : Hayflick Limit (1) .svg, par Azmistowski17, via Wikimédia Commons, CC-BY-SA-4.0, https://commons.wikimedia.org/wiki/File:Hayflick_Limit_(1).svg
Il existe une enzyme permettant de maintenir la taille des télomères et permettant ainsi des divisions infinies. Elle ne s’exprime que dans les cellules souches de la moelle osseuse et dans les cellules germinales à l’origine des spermatozoïdes des ovules. C’est la « télomérase » codée par le gène TERT situé sur le chromosome 5.
Document 2 : Télomères et télomérase
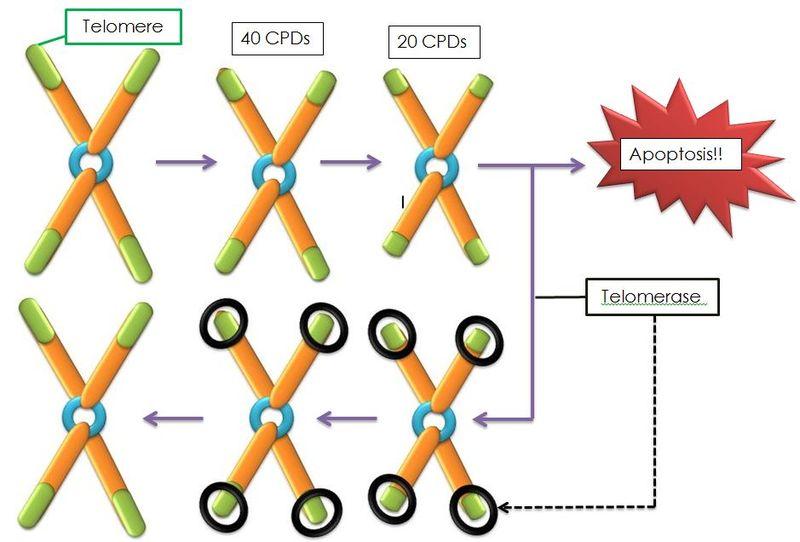
Sources : Telomerase & Telomeres.JPG, par
DéveloppementalBiologie, via Wikimédia Commons, CC-BY-SA-3.0, https://commons.wikimedia.org/wiki/File:Telomerase%26Telomeres.JPG
La télomérase rajoute tout d’abord des nucléotides sur un fragment de la double hélice puis l’enzyme habituelle (l’ADN polymérase) synthétise le brin complémentaire pour reconstituer un télomère complet. Les divisions sont alors à nouveau possibles.
Document 3 : Action de la télomérase
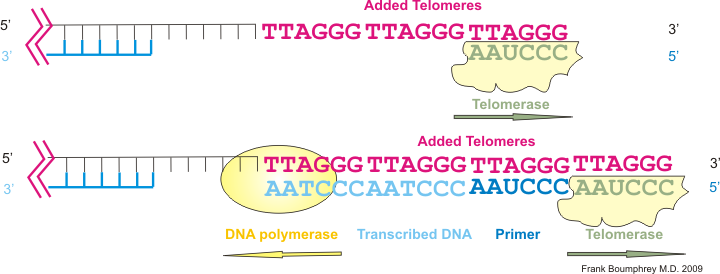
Sources : Telomerase & Telomeres.JPG, par Boumphreyfr, via Wikimédia Commons, CC-BY-SA-3.0, https://commons.wikimedia.org/wiki/File:Telomerase.png
Dans les cellules cancéreuses, la télomérase est particulièrement active ce qui explique la croissance rapide et incontrôlée des cellules malgré l’inhibition de contact normalement enclenchée par la juxtaposition des cellules voisines. Cette forte activation vient d’une mutation de la séquence régulatrice du gène TERT situé sur le chromosome 5 et codant pour la télomérase. Pour que le facteur de transcription ETS1 se fixe sur la séquence régulatrice, précisément sur le promoteur, permettant ainsi à l’ARN polymérase de se fixer et de transcrire le gène TERT à l’origine de la télomérase, il faut qu’il y ait dans cette séquence régulatrice une succession de 4 nucléotides spécifiques : CCTT. Suite à une mutation, chez un individu atteint d’un cancer, on a observé qu’une cytosine est remplacée par une thymine en position 146 avant le gène, créant ainsi une succession de type CCTT faisant apparaître un autre site de fixation du facteur de transcription ETS1. Ainsi dans des cellules somatiques qui ne devraient pas reconstituer leurs télomères, la télomérase est surexprimée, les télomères réparés, les divisions infinies : un clone apparaît formant une masse de cellules anormale appelée tumeur.
Document 4 : Régulation du gène TERT

II La vie d’une cellule est normalement écourtée en cas de mutation
Il existe plusieurs points de contrôle du cycle cellulaire. Un des points de contrôle se situe avant la duplication et consiste à vérifier l’état de l’ADN et à enclencher si besoin une réparation de celui-ci. Ce système empêche une cellule anormale de se développer. Ce point de contrôle est activé quand la lésion de l’ADN est repérée, et ceci pour éviter que les erreurs ne se propagent dans les cellules-filles. Une enzyme appelée CdK2 associée à une cycline (cycline E) est responsable du passage de la phase G1 à la phase GS de la division cellulaire. Elle possède une action sur les enzymes impliquées dans la réplication.
Document 5 : Points de contrôle du cycle cellulaire

Sources : Schéma cycle cellulaire.GIF, par Valérie Villeneuve, via Wikimédia Commons, CC-BY-SA-3.0,2.5,2.0,1.0, https://commons.wikimedia.org/wiki/File:Sch%C3%A9ma_cycle_cellulaire.GIF
Si l’ADN est abîmé il faut empêcher le passage de la phase G1 à la phase S et donc inactiver la CdK2-cycline. Une enzyme appelée ATM est alors recrutée. Elle va phosphoryler l’inhibiteur MDM2 de la protéine p53. Cette dernière libérée va jouer son rôle de facteur de transcription. Elle se fixe sur le promoteur du gène de la protéine p21 inhibitrice de la CdK2-cycline. Ainsi il n’y a pas passage de la phase G1 à S : le cycle est bloqué et l’ADN peut être réparé.
Document 6 : Recrutement et activation de la protéine p53

Sources : protéines cibles ATM (nouveau) .png, Biochimie cellulaire II ( discussion ), via Wikimédia Commons, CC-BY-SA-3.0, modifié par Sandra Rivière, https://commons.wikimedia.org/wiki/File:ATM_target_proteins_(new).png
Document 7 : Action de la protéine p53
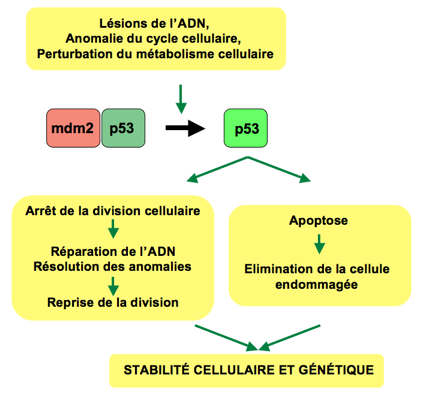
Source : P53 Pathway.png par Thierry Soussi via Wikimédia Commons, domaine publique, https://commons.wikimedia.org/wiki/File:P53_Pathway.png
Si la protéine p53 est mutée dans la partie qui se fixe au promoteur du gène de la protéine p21, l’ARN polymérase ne pourra pas se fixer sur celui-ci et cette dernière ne sera pas produite. La CdK2-cycline fonctionnera alors en permanence. Le cycle cellulaire deviendra anarchique et ne s’arrêtera jamais, provoquant l’apparition d’une masse de cellules : un clone de cellules associées stables est formé. On parle de dysplasie. Si la cellule de départ présentait une mutation empêchant un métabolisme normal, alors l’organe touché deviendra déficient : on parle de cancer.
Document 8 : Mise en place d’un cancer

III Un organisme présente forcément des mutations
Les cellules somatiques constituant les tissus sont des clones. Elles ont donc toute la même information génétique. Une erreur de réplication dans l’une d’elles va être à l’origine d’une mutation. En connaissant le taux moyen de mutations d’une cellule somatique par division, il est possible d’estimer le nombre théorique de mutations dans un organisme humain lors de son développement.
Les taux d’erreur de l’ADN polymérase et de 1/1 000 000 000 soit 10-9. Un homme d’un mètre 70 et pesant 70 kg, est constitué d’environ 3 × 1013 cellules.
On peut dans un premier temps calculer le nombre de divisions qui ont été nécessaires pour former cet individu. Ainsi si on part d’une cellule pour obtenir quatre cellules, il y a eu matériellement trois divisions. La première division a donné 2 cellules puis chacune de ces cellules s’est divisée pour en donner 2 autres. Il y a donc eu 1 + 2 = 3 divisions pour obtenir 4 cellules soit 4 – 1 (nombre de cellules à l’arrivée moins la cellule de départ). Ainsi le nombre de divisions nécessaires pour obtenir un organisme à n cellules sera donc de n-1.
Pour l’individu cité, le nombre de divisions nécessaires à son édification est de (3 × 1013 – 1)
Or le taux de mutations étant de 10-9, le nombre total de mutations présentes chez cet individu est de (3 × 1013 – 1) x 10-9 soit environ 3 × 104.
Cette valeur n’est pas totalement exacte puisque ne sont pas pris en compte les mitoses réalisées pour remplacer les cellules qui meurent et qui sont renouvelées régulièrement. Il y a donc eu chez cet individu, un nombre de mutations bien supérieures à 3 × 104.
IV Un exemple de phénotype lié à une mutation de la séquence codante d’un gène
Les mutations somatiques conservées dans les cellules des tissus issues de divisions cellulaires finissent par disparaître à la mort de l’individu.
Les mutations touchant les cellules germinales sont transmises si la cellule touchée participe à la fécondation. Dans ce cas la mutation provoquera l’apparition d’un caractère héréditaire.
Une mutation sur une séquence d’un gène a une conséquence aux échelles moléculaire, cellulaire et macroscopique.
La séquence d’un gène est responsable d’une séquence en acides aminés. Cette dernière déterminera la conformation spatiale et la fonction de la protéine. Dans le cas d’une maladie génétique, une mutation d’un gène considéré entraîne la modification de la structure spatiale de la protéine la rendant non fonctionnelle. La cellule touchée est alors non fonctionnelle et l’individu est malade.
La phénylcétonurie est une maladie génétique rare, liée à un déficit en phénylalanine hydroxylase, entraînant l’accumulation de phénylalanine dans le sang et le cerveau. La phénylcétonurie est une maladie traitable et lorsque le régime pauvre en phénylalanine est conduit de manière optimale, il permet un développement intellectuel normal.
La phénylalanine hydroxylase (PAH) est une enzyme fabriquée suivant un programme génétique très précis. Une mutation du gène de structure porté par le chromosome 12, peut entraîner la formation d'une protéine à activité réduite ou nulle. Cette enzymopathie est une maladie héréditaire récessive autosomique. Elle n'apparait que si les deux parents sont hétérozygotes et le risque à chaque grossesse d'avoir un enfant atteint est de 25%. L'incidence est d'environ 1/16000 naissances, ce qui correspond à environ 1 hétérozygote sur 60 dans la population. Le gène de la phénylalanine hydroxylase est très polymorphe : cela signifie qu’il existe plusieurs allèles pour ce gène.
L’allèle de référence est l’allèle « phenorm ». L’allèle « phe 6 » permet un phénotype sain alors que l’allèle « phe 3 » aboutit à une phénylcétonurie sévère et l’allèle « phe 4 » à une phétylcétonurie moyennement sévère.
La comparaison sous ANAGENE des allèles révèle pour l’allèle phe3 en position de nucléotide 331, la substitution d’une cytosine par une thymine provoquant l’apparition d’un codon stop en position 111 de la chaîne d’acides aminés. La protéine est alors incomplète on parle de mutation non-sens.
Document 9 : Comparaison simple des allèles de la PAH, l’accent est porté sur l’allèle « phe3 »

Document 10 : Comparaison simple des séquences en acides aminés de la PAH, l’accent est porté sur la protéine « phe3 »

Pour l’allèle phe4 on remarque au nucléotide 473, la substitution d’une guanine par une adénine provoquant l’apparition d’une glutamine à la place d’une arginine en position 158 de la chaîne d’acides aminés. La protéine est modifiée mais active. On parle de mutation faux-sens.
Pour l’allèle phe6, au nucléotide 696, il y a la substitution d’une adénine par une guanine. Ceci ne provoque pas de modification d’acides aminés. On parle de mutation silencieuse car il n’y alors pas de changement de la conformation spatiale de la protéine.
Ainsi on distingue ainsi trois catégories de mutation :
- les mutations silencieuses qui ne se voient pas et dont la cause provient de la redondance du code génétique. Le codon muté code pour un acide aminé identique : ainsi la séquence en acides aminés de la protéine restera identique et le phénotype macroscopique ne sera pas modifié.
- la mutation faux-sens : elle code pour un sens légèrement différent du sens de départ. La mutation provoque le remplacement d’un acide aminé par un autre et la protéine peut perdre partiellement ou totalement sa fonction. L’individu est alors malade mais peut présenter une forme atténuée de la maladie.
- La mutation non-sens est une mutation qui n’a pas de sens ! Elle ne code pour rien ! L’apparition d’un codon stop provoque l’arrêt de la synthèse de la protéine. La protéine est absente et sa fonction n’est pas assurée chez l’individu qui sera alors malade.
Document 11 : Différentes catégories de mutations

Source : Point CAA.png par Lucquessoy, via Wikimedia commons, : CC-BY-SA-3.0 https://commons.wikimedia.org/wiki/File:Point_CAA.png
V Un exemple de phénotype lié à une mutation de la séquence régulatrice d’un gène
Parfois certaines mutations concernent les séquences régulatrices de gènes comme les promoteurs.
Les cellules qui se différencient n’expriment qu’une partie de leur génome. Elles acquièrent ainsi une identité par les gènes qu’elles expriment. Il existe un processus de contrôle de l’expression des gènes par l’intermédiaire d’un facteur de transcription qui se fixe directement sur l’ADN au niveau de séquence régulatrice du promoteur du gène. À ce niveau le facteur de transcription ouvre la double hélice d’ADN pour permettre la fixation de l’ARN polymérase et donc la transcription et l’expression du gène. Sans promoteur, la protéine codée par le gène ne peut être produite. Cette séquence se situe toujours en amont ou à côté du gène et donc ne fait pas partie de la séquence codante mais elle en permet l’expression et donc sa transcription puis sa traduction en protéine. Sans promoteur la protéine n’est pas synthétisée.
Prenons l’exemple d’une mutation dans une lignée germinale sur la séquence régulatrice d’un gène impliqué dans une bêta thalassémie. Travaillons sur la production d’hémoglobine au cours du développement d’un individu. Un individu adulte possède une hémoglobine normale constituée de quatre chaînes polypeptidiques identiques deux à deux : 2 chaînes alpha et 2 chaînes bêta.
Document 12 : Molécule d’hémoglobine observée sous Rastop
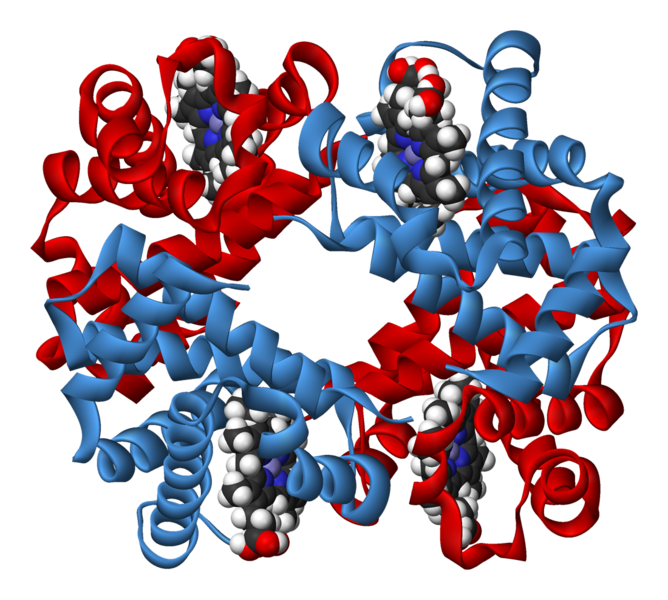
Source : Hemoglobin-3D-ribbons.png, par Benjah-bmm27 via Wikimédia Commons, domaine publique, https://commons.wikimedia.org/wiki/File:Haemoglobin-3D-ribbons.png
Il existe un seul gène codant pour la globine bêta sur le chromosome 11. On a recensé de multiples mutations qui aboutissent à des formes plus ou moins graves de la maladie. Il a été constaté que parfois un individu n’exprime pas de globine bêta alors qu’il possède deux allèles codant non mutés.
La bêta thalassémie est une maladie héréditaire récessive. Il existe plus de 200 anomalies génétiques atteignant le gène bêta, la plupart sont des mutations ponctuelles. Selon la localisation de la mutation sur le gène, on observe soit une absence totale d’expression et donc l’absence de globine bêta soit une diminution plus ou moins forte de cette expression est donc une diminution plus ou moins forte de la synthèse de la globine bêta.
Dans les formes graves, de la globine bêta n’est pas produite alors que l’alpha est produite normalement. Les chaînes alpha en excès s’associent en tétramère qui sont instables et précipitent dans le cytoplasme des cellules sanguines, provoquant leur apoptose (mort des cellules). Le malade présente alors une anémie en globules rouges et son tissu osseux essaie de produire de plus en plus de globules rouges. Ce tissu osseux va alors augmenter de volume entraînant une déformation squelettique parmi d’autres symptômes. Ce cas nécessite une transfusion à vie pour corriger l’anémie et supprimer la production intensive de globules rouges.
Document 13 : Évolution des proportions normale d’alpha globine et de bêta globine dans le sang avant et après la naissance.
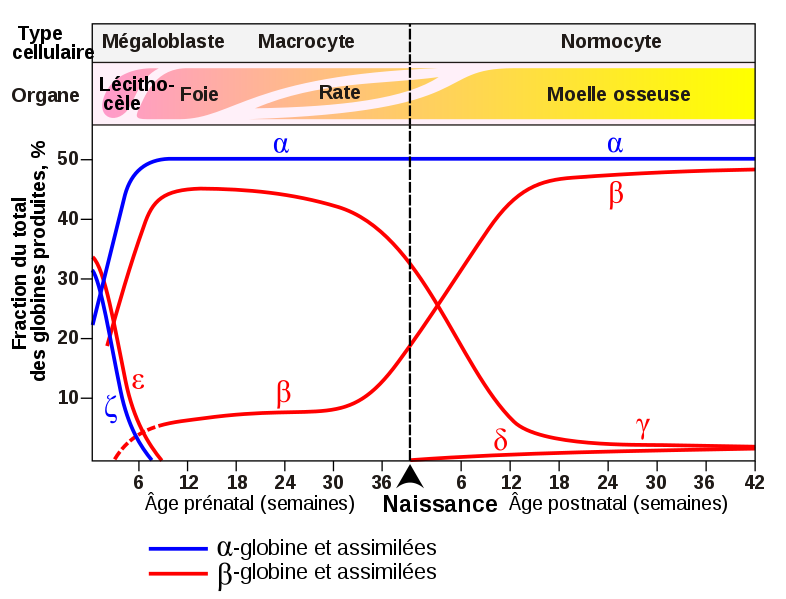
Source : Globines humaines à la naissance.svg, par Léonid 2 via Wikimédia Commons, domaine publique, https://commons.wikimedia.org/wiki/File:Globines_humaines_%C3%A0_la_naissance.svg
Si l’individu reçoit deux allèles de bêta globine mutés sur le promoteur, ces derniers ne peuvent pas s’exprimer. Seule la globine alpha est produite. Dans ce cas, à partir de 36 mois on observe chez l’enfant une hémoglobine instable et une apoptose prononcée des globules rouges provoquant une anémie.
VI Un exemple de phénotype lié l’accumulation de mutations
On peut observer des effets phénotypiques de l’accumulation de mutations somatiques.
La leucémie myéloïde chronique fait partie des maladies du sang regroupées sous le nom de « syndrome myéloprolifératifs ». Elle est caractérisée par une production excessive et persistante au sein de la moelle osseuse de globules blancs. Une partie des globules blancs anormaux sont des cellules immatures dont le développement n’est pas fini quand elles passent dans le sang. Cette maladie est liée à l’apparition d’une anomalie liée à la translocation réciproque de portions des deux chromosomes dans les cellules souches de la moelle osseuse. Il y a alors apparition d’un petit chromosome 22 anormal appelé chromosome Philadelphie du nom de la ville ou travaillent les deux chercheurs qui l’ont découvert en 1960.
Il y a assemblage par erreur d’un gène du chromosome 9, le gène ABL avec un gène du chromosome 22, le gène BCR, créant le gène dit BCR-ABL présent uniquement dans les cellules anormales. Le gène ABL produit une enzyme la tyrosine kinase régulable permettant la production des globules blancs. Le gène BCR-ABL produit une tyrosine kinase non régulable provoquant ainsi une surproduction de globules blancs. Cette anomalie génétique est acquise par des cellules souches anormales et n’est donc pas héréditaire. Les causes de son apparition sont inconnues.
Document 14 : Formation du chromosome Philadelphia
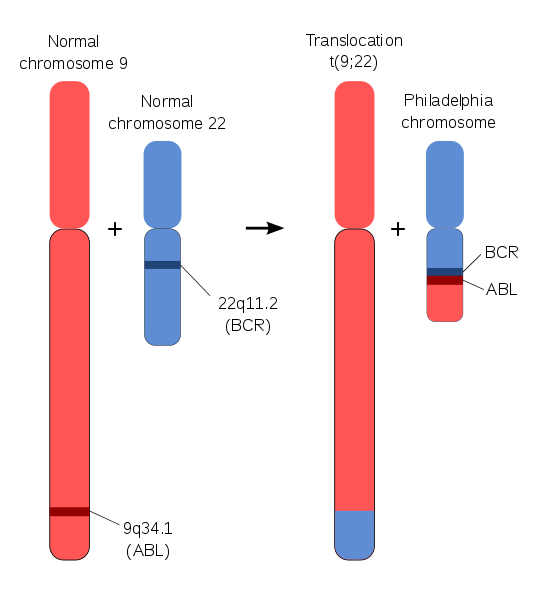
Source : Schéma du chromosome de Philadelphie.svg, par Aryn89 via Wikimedia commons, CC-BY-SA-4.0, https://commons.wikimedia.org/wiki/File:Schematic_of_the_Philadelphia_Chromosome.svg
La leucémie myéloïde chronique évolue en trois phases. La première phase est la phase chronique et c’est en général à ce stade que la maladie est diagnostiquée. La leucémie évolue lentement et il n’y a pas de symptômes. Les analyses du sang et de la moelle osseuse révèlent peu de globules blancs anormaux.
Le deuxième stade est la phase d’accélération qui correspond à une augmentation de la proportion de globules blancs anormaux dans le sang et dans la moelle osseuse ainsi qu’une élévation du nombre de cellules présentant le gène BCR-ABL ou encore d’autres anomalies chromosomiques. Les symptômes apparaissent non spécifiques et sont fréquents : fatigue, perte d’appétit, fièvre…. La maladie évolue après plusieurs mois vers la phase aiguë où la moelle osseuse est envahie par les globules blancs anormaux.
C’est donc l’élévation de la charge en cellules présentant le gène BCR-ABL qui est à l’origine de l’augmentation des globules blancs anormaux. Cette élévation de charge en cellules anormales correspond donc à l’évolution d’un clone muté au sein des cellules souches. Il y a eu absence de réparation de l’anomalie génétique ayant permis de la conserver.
Document 15 : Cellules touchées par l’anomalie génétique responsable de la leucémie myéloïde chronique.

{kind=link}
{kind=link}
Source : Diagramme montrant les cellules dans lesquelles AML démarre CRUK 297 fr.svg , par Cancer Research UKvia Wikimédia Commons, domaine publique, https://commons.wikimedia.org/wiki/File:Diagram_showing_the_cells_in_which_AML_starts_CRUK_297_fr.svg
Lignée clonale -SVT - LA VIE Term spé #3 - Mathrix
Date de dernière mise à jour : 13/06/2021